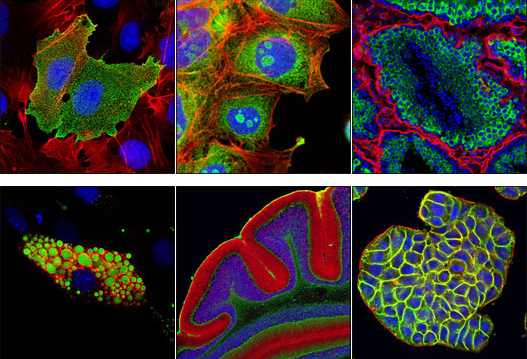
没有一个免疫荧光(IF)流程是真正完美的。要有一个漂亮的能用于发表的IF结果,需要根据样本类型、目的蛋白、抗体、抗体操作和抗原抗体结合进行每一步的优化。
细胞样本制备或者染色的问题均可导致不理想的结果。要进行优化,就应了解IF染色的基本知识,这样可节省时间,并且离漂亮的染色更近一步。
下面建议是根据Proteintech厂家的The Complete Guide To Optimizing Immunofluorescence Staining手册进行编译的,主要针对细胞免疫荧光。具体的实验还需要特定的调整。
一、细胞培养
染色时,细胞密度应在60~80%。密度过高或过低均可影响细胞结构和形态。密度过低也不易有合适的成像区域。可在12孔或24孔板内放置玻片以接种细胞。加适量培养基让细胞生长,直至达到合适的细胞密度。如果做细胞刺激实验,则需要在刺激实验后重新细胞计数,并预计细胞达到合适密度所需的时间。

提示:应了解你的目的蛋白。例如缝隙连接蛋白的染色需要合适的细胞密度,不同细胞密度可能导致结果不同。
二、玻片
玻片应高压消毒,在无菌台上用乙醇洗涤,并在完全干燥后再使用。可用镊子把玻片放到培养板内。
三、包被
一些细胞不贴在玻片上。在这种情况下,可对玻片进行包被。选择哪种包被基质依赖于细胞类型。大多数细胞系很好地黏附在多聚赖氨酸上。其他基质蛋白有层粘连蛋白(ESC等)、胶原(原代角质细胞/肝细胞等)、明胶(微血管内皮细胞等)、或纤粘连蛋白(NSC等)。包被应均匀,并根据说明书确定包被条件(时间、温度),之后用1x PBS或细胞培养级别的无菌水彻底洗净表面。
四、固定
当细胞达到理想密度时,可吸取培养基,用PBS洗2遍后加固定液固定,再用PBS洗2遍。如不立即染色,留一些PBS在孔内,4°C密闭保存。固定的细胞可以在阴凉处保存几周。要确保玻片没有完全变干。
五、自发荧光
自发荧光使IF复杂化。确定自发荧光的来源很重要。有生物性自发荧光和固定剂导致的自发荧光。生物性自发荧光可来自线粒体、溶酶体和含有芳香族氨基酸的组分(如胶原),其中最显著是黄素类的辅酶如FAD、FMN,或吡啶核苷酸如NADH。FAD在450nm处有吸收,515nm处发射。NADH在340nm处有吸收,515nm处发射。一些醛类固定剂也会和蛋白交联,形成自发荧光物质,这时可还原醛基为羟基来消除自发荧光问题(如使用硼氢化钠)。注意市面上的商品化自发荧光消除剂避免使用时过量,因为过量常常导致目的信号的丢失。
以下为常用固定剂的比较
|
固定剂类型 |
名称 |
优点 |
缺点 |
|
有机溶剂 |
甲醇 |
细胞骨架完好 |
对抗原表位有损害。脂质组分丢失 |
|
有机溶剂 |
丙酮 |
对抗原表位作用较温和 |
脂质组分丢失 |
|
化学交联剂 |
多聚甲醛 |
细胞形态良好 |
抗原表位交联,有自发荧光 |
六、封片剂
抗淬灭剂有利于保留荧光信号。在选择抗淬灭剂时,应考虑该该样本是否想长期保存,还是只临时观察?是哪种荧光素标记?是否为多重染色?气泡、洗片不充分、抗体孵育过度、固定过度都会导致假阳性。抗淬灭剂过多或过少也会产生假信号或样本损害。
使用对照有利于辨别非特异染色。如:
l 样本不染色以判断是否有自发荧光。
l 省略一抗而只用二抗,来判断二抗是否为特异结合。
l 用封闭肽阻断一抗,以判断一抗是否为特异性结合。
l 对于多重染色,也可分别单染,以确定不存在交叉反应。

七、IF常见问题
无染色或染色弱
|
可能原因 |
推荐尝试 |
|
抗体条件未优化 |
采用抗体浓度梯度来优化条件,一抗室温孵育或4度过夜 |
|
目的蛋白低表达 |
采用信号级联放大(不要用荧光直接法) |
|
抗原表位破坏 |
原因是固定过度,应减轻固定,或换固定剂 |
|
抗体不适合IF检测天然蛋白 |
可做非变性的Western Blot并观察结果 |
存在非特异染色/无特异染色
|
可能原因 |
解决方案 |
|
目的蛋白为核蛋白 |
样本应破膜处理 |
|
荧光信号弱或者没有 |
在暗处保存荧光标记抗体。采用直标抗体。采用信号增强剂 |
|
荧光淬灭 |
在暗处保存荧光标记抗体。换用抗淬灭剂 |
|
人工假象 |
可能与下列因素有关:细胞培养、细胞密度、洗片不充分、固定过度、封片剂 |
存在背景染色
|
可能原因 |
解决方案 |
|
一抗或二抗非特异结合 |
使用对照。延长封闭时间 |
|
样本洗涤不充分 |
增加洗片次数或延长洗片时间 |
|
抗体孵育温度太高 |
4度孵育 |
|
不适当的固定导致假象或抗原破坏 |
减少固定步骤,换固定剂 |
|
破膜导致细胞和蛋白损害 |
减少或跳过破膜步骤 |
|
玻片过干 |
总是保持玻片潮湿 |
