Our cell separation columns are designed as a rapid, one step tool to enrich for specific cell populations. We have attempted to make the columns simple and user friendly. To ensure that all users get maximal efficiency from these columns, we have summarized some common problems below. We attempt to identify the likely sources of these problems and possible courses of action to be taken to avoid them. We urge users to review these tips and if additional advice is required to contact our technical service department.
Possible Source Test or Action Air lock in column tip Tap side of column to dislodge air bubble
Possible Source Test or Action Cell clumps and debris have accumulated on the top white filter Remove cell debris that has accumulated on the white filter with a sterile pipette Reduce the amount of time single cells incubate in a small volume as it may reduce the tendency of cells to form clumps
Possible Source Test or Action Poor specimen preparation Ensure that suspension contains single cells with no clumps and minimal RBC Column was either overloaded or underloaded Load the optimal number of total cells indicated in the product insert Improper calculation Percent recovery should be calculated by dividing the number of target cells recovered by the number of target cells loaded
Possible Source Test or Action Non-optimal number of total cells loaded Reduce the total number of cells loaded and determine by immunophenotype if the frequency of the contaminating population is reduced Poor specimen preparation or tissue source High frequency of dead cells in recovered sample. Determine cell viability and perform immunophenotype analysis gating only on live cells
ELISA Development
For further assistance, please contact our technical service department.
- High background
- No signal
- Too much signal - whole plate turned uniformly blue
- Standard curve achieved but poor discrimination between points
- Poor duplicates
- Poor assay to assay reproducibility
- No signal when a signal is expected, but standard curve looks fine
- Samples are reading too high, but standard curve looks fine
- Very Low Readings Across the Plate
- Green color develops upon addition of stop solution when using streptavidin-HRP
- Edge Effects
- Drift
Problem: High Background
Possible Source Test or Action Insufficient washing See washing procedure on page 4 of theELISA Development Guide Increase number of washes Add a 30 second soak step inbetween washes
Problem: No signal when a signal is expected
Possible Source Test or Action Reagents added in incorrect order, or incorrectly prepared Repeat assay Check calculations and make new buffers, standards, etc.
Review protocolContamination of HRP with Use fresh reagents Not enough antibody used Increase concentration Standard has gone bad (if there is a signal in the sample wells) Check that standard was handled according to directions.
Use new vial.Buffer containing FCS used to reconstitute antibodies Requalify your reagents of choice Capture antibody did not bind to plate Use an ELISA plate (not a tissue culture plate)
Dilute in PBS without additional proteinBuffers contaminated Make fresh buffers
Problem: Too much signal - whole plate turned uniformly blue
Possible Source Test or Action Insufficient washing/washing step skipped - unbound peroxidase remaining See washing procedure on page 4 of theELISA Development Guide Substrate Solution mixed too early and turned blue Substrate Solution should be mixed and used immediately Too much streptavidin-HRP Check dilution, titrate if necessary Plate sealers or reagent reservoirs reused, resulting in presence of residual HRP. This will turn the TMB blue non-specifically Use fresh plate sealer and reagent reservoir for each step Buffers contaminated with metals or HRP Make fresh buffers
Problem: Standard curve achieved but poor discrimination between points (low or flat curve)
Possible Source Test or Action Not enough streptavidin-HRP Check dilution, titrate if necessary Capture antibody did not bind well to plate Use an ELISA plate (not a tissue culture plate)
Dilute in PBS without additional proteinNot enough detection antibody Check dilution, titrate if necessary Plate not developed long enough Increase Substrate Solution incubation time
Use recommended brand of Substrate SolutionIncorrect procedure Go back to General ELISA Protocol; eliminate modifications, if any Improper calculation of standard curve dilutions Check calculations, make new standard curve
Problem: Poor Duplicates
Possible Source Test or Action Insufficient washing See washing procedures on page 4 of theELISA Development Guide
If using an automatic plate washer, check that all ports are clean and free of obstructions, add a 30 second soak step and rotate plate halfway through the washUneven plate coating due to procedural error or poor plate quality (can bind unevenly) Dilute in PBS without additional protein
Check coating and blocking volumes, times and method of reagent addition. Check plate used
Use an ELISA plate (not a tissue culture plate)Plate sealer reused Use a fresh plate sealer for each step No plate sealers used Use plate sealers Buffers contaminated Make fresh buffers
Problem: Poor assay to assay reproducibility
Possible Source Test or Action Insufficient washing See washing procedure on page 4 of theELISA Development Guide
If using an automatic plate washer, check that all ports are clean and free of obstructionsVariations in incubation temperature Adhere to recommended incubation temperature
Avoid incubating plates in areas where enviromental conditions varyVariations in protocol Adhere to the same protocol from run to run Plate sealer reused, resulting in presence of residual HRP which will turn the TMB blue Use fresh plate sealer for each step Improper calculation of standard curve dilutions Check calculations, make new standard curve
Use internal controlsBuffers contaminated Make fresh buffers
Problem: No signal when a signal is expected, but standard curve looks fine
Possible Source Test or Action No cytokine in sample Use internal controls
Repeat experiment, reconsider experimental parametersSample matrix is masking detection Dilute samples at least 1:2 in appropriate diluent, or preferably, do a series of dilutions to look at recovery
Problem: Samples are reading too high, but standard curve looks fine
Possible Source Test or Action Samples contain cytokine levels above assay range Dilute samples and run again
Problem: Very low readings across the plate
Possible Source Test or Action Incorrect wavelengths Check filters/reader Insufficient development time Increase development time Coated plates are old and have gone bad Coat new plates Capture antibody did not bind to the plate Use an ELISA plate (not a tissue culture plate)
Dilute in PBS without additional proteinBuffer containing FCS used to reconstitute antibodies Requalify your reagents of choice
Problem: Green color develops upon addition of stop solution when using streptavidin-HRP
Possible Source Test or Action Reagents not mixed well enough in wells Tap plate
Problem: Edge Effects
Possible Source Test or Action Uneven temperatures around work surface Avoid incubating plates in areas where environmental conditions vary Use plate sealers
Problem: Drift
Possible Source Test or Action Interrupted assay set-up Assay set-up should be continuous - have all standards and samples prepared appropriately before commencement of the assay Reagents not at room temperature Ensure that all reagents are at room temperature before pipetting into the wells unless otherwise instructed in the antibody inserts
Possible Source Test or Action Interrupted assay set-up Assay set-up should be continuous - have all standards and samples prepared appropriately before commencement of the assay Reagents not at room temperature Ensure that all reagents are at room temperature before pipetting into the wells unless otherwise instructed in the antibody inserts
ELISA failure can be attributed to many factors. Many errors can be avoided if the protocol is read and fully understood before starting the assay. On identifying assay failure, check the expiration dates of the individual reagents and ensure that all the reagents have been stored as indicated on the product label. Once this has been established, check for signs of instability or deterioration in reagent solutions, (e.g., precipitation or discoloration). All substrate solutions should be colorless. Whenever possible, use clean plastic disposable pipettes, tips, and containers for reagent preparation and storage. Avoid cross-contamination of kit reagents by changing pipette tips between addition of each standard, sample and reagent. Finally, ensure that specified incubation times and temperatures have been adhered to and that no substitution of kit reagents has occurred. To improve accuracy, it is recommended that samples and standards be run in duplicate. For further assistance, please contact our technical service department.
Possible Source Test or Action Incomplete washing of wells Ensure that wash apparatus is working correctly Inadequate aspiration of wells Wells should appear dry after aspiration Unequal mixing of reagents Ensure adequate mixing Pipetting error Repeat assay
Possible Source Test or Action Improper dilution of highest standard, standard curve, or blank values* Ensure use of appropriate diluent as blank value. Ensure accurate completion of 2-fold dilution series* ** Incomplete washing of wells Ensure that wash apparatus is working correctly Inadequate aspiration of wells Wells should appear dry after aspiration Unequal volumes added to wells Check pipette function; recalibrate if necessary Color Reagents A and B mixed too early* Substrate Solution must be used within 15 minutes of mixing* Pipetting error Repeat assay
Possible Source Test or Action Inadequate volume of substrate added to wells Check pipette function Incomplete mixing of Color Reagents A and B* Ensure that equal volumes of Color Reagents A and B are properly mixed* Incorrect incubation times or temperature Adhere to recommended incubation times and temperatures Conjugate or color reagent failure Check by mixing equal volumes of Color Reagents A and B and conjugate. Color should develop immediately*
Possible Source Test or Action Uneven temperatures around work surface Adhere to recommended incubation periods and temperatures Inadequate fixing of plate cover, leading to evaporation Ensure correct use of plate cover
Possible Source Test or Action Interrupted assay set-up or reagents not at room temperature Assay set-up should be continuous. Have all standards/samples prepared appropriately before commencement of the assay
Possible Source Test or Action We as manufacturers take great care to ensure that our products are suitable for use with all valid samples, as designated in the product inserts. However, it is possible that in some cases high levels of interfering factors may cause unusual results.
ELISpot
For further assistance, please contact our technical service department.
Observation Problem Corrective Action Following the incubation with BCIP/NBT chromogen and rinsing the microplate with deionized water, the dark-blue background color of filter membrane attenuates visualization and quantitation of spots. Wet membrane Microplates cannot be analyzed accurately until PVDF filter membranes are completely dry. Wait until membrane becomes dry, usually 15 - 30 minutes at 37° C or 60 - 90 minutes at room temperature The number of spots in the wells that contained the cells is high but their contrast as well as intensity of staining in the Positive Control wells is low. Underdevelopment - may be a result of using Streptavidin-AP and/or BCIP/NBT solutions that have not been brought to room temperature Bring reagents to room temperature before adding to the wells. The number of spots in the wells that contained cells is lower than expected whereas Positive Control wells turned black-blue. Cell stimulation problem Ensure that reagents used to stimulate the cytokine release from the cells retained their biological activity. One way to check is to perform immunocytochemistry on fixed cells after stimulation. Too few cells added to the wells Increase the number of cells added per well. Following incubation with BCIP/NBT and drying the microplate, the density of the spots makes it difficult to quantify them. Too many cells were added to the wells Make dilutions of cells (i.e., 1 x 106, 5 x 105, 1 x 105, 5 x 104, 1 x 104 cells per well) to determine the optimal number of cells that will result in formation of distinct spots.
Fluorokine® Receptor Detection Kits are designed as alternative reagents for the detection of cell surface cytokine receptors by flow cytometry. Although the staining procedure of cells with this line of reagents is straight forward, some situations can present difficulties in data interpretation. In the table below, we attempt to point out some common problems that users of Fluorokine Receptor Detection Kits may encounter. The variable nature of cytometry instrumentation and instrument set-up can dramatically influence the quality of data generated with these reagents.
Cell surface receptor analysis involves the monitoring of protein structures that are in a dynamic equilibrium with their environment so changes in the medium such as pH, temperature, ionic strength, or presence of other proteins and/or cytokines will ultimately influence the binding properties of each labeled ligand. Lastly, the biological activity of labeled ligands can modulate a cell’s receptor density. We hope the tips below will help investigators overcome simple obstacles, but if additional help is required, we encourage you to contact our technical service department.
Possible Source Test or Action Cell processing error Check all steps of the staining protocol to ensure proper cell processing Cells were left on ice too long and receptors may have been downregulated Stain fresh cells Cells may have lost or decreased receptor numbers during culture Use another receptor positive cell line to test reagent or retest same cells at different cell cycle stages
Possible Source Test or Action Voltage being applied to photomultiplier tubes on cytometer are set too high Reduce PMT voltage until >95% of events fall in the first decade of the log scale Concentration of Fluorokine in the reaction is too high Dilute the Fluorokine and compare signal intensity against cells that do express receptors Transfected cells have upregulated other cell surface structures Test Fluorokine on non-transfected cells or on other transfectants using a different parent line
Possible Source Test or Action Cells were not pre-treated with Ig to block Fc receptors Repeat the test by first treating cells with mouse or human Ig to block Fc receptors
Possible Source Test or Action Fluorokine present at too high concentration Repeat reaction using less Fluorokine Addition of unconjugated cytokine upregulated receptors Perform all reactions at 2 - 8° C and in the presence of metabolic blockers, e.g.: azide, cytochalasin B, etc. Numerically the inhibition is not 100% Comparison of mean fluorescence intensity between the two conditions may reveal substantial inhibition
Immunohistochemistry
Technically, IHC and ICC are relatively simple and straightforward experimental methods. However, there are many variables which must be identified and optimized for each individual IHC/ICC study. Optimization of IHC/ICC may also require troubleshooting a variety of factors. The tables below highlight common IHC/ICC issues and provide appropriate experimental actions. For further assistance, please contact our technical service department.
Problem: Lack of Staining
Possible Source Test or Action Lack of antigen. Check protein expression by in situhybridization (in some rare cases translation may be blocked even though mRNA is detected). Antibodies do not work due to improper storage. Follow storage instructions on the datasheet. In general, aliquot antibodies into smaller volumes sufficient to make a working solution for a single experiment. Store aliquots in a manual defrost freezer (-20 to -70 °C) and avoid repeated freeze-thaw cycles. Inactive primary or secondary antibodies. Test reporter system independently to assess reagent viability. Inadequate tissue fixation. Try increasing the fixation time or try a different fixative. Tissue overfixation. Reduce the duration of the immersion or post-fixation steps. If immersion fixation cannot be avoided (for example, collection ofpostmortem tissues or biopsies in pathology lab), antigens may be unmasked by treatment with antigen retrieval reagents. Incompatible secondary and primary antibodies. Use a secondary antibody that will interact with the primary antibody. For example, if the primary antibody was raised in rabbits, use an anti-rabbit secondary antibody. Antigen was destroyed before incubation with the primary antibody. If quenching of endogenous peroxidase was done prior to the addition of primary antibodies, block peroxidase after incubation with the primary antibody. Epitope altered during fixation or embedding procedure. Try restoring immunoreactivity through various antigen retrieval techniques.
Embed tissue at 58 °C or below.Antigen retrieval was ineffective. Increase the time of treatment or change the treatment solution. Reagents omitted or used in wrong order. Repeat staining and confirm that correct reagents are used and are added in the correct order. Problem: High Background
Possible Source Test or Action High concentration of primary and or secondary antibodies. Titer antibody to determine optimal concentration needed to promote a specific reaction of the primary and the secondary antibodies. Hydrophobic interactions of the antibody and proteins in the tissue. Lower the ionic strength of the antibody diluent (particularly monoclonal antibodies respond well to reducing the salt concentration). Non-speci?c binding of primary and/or secondary reagents to tissues. Use blocking step just prior to primary antibody incubation (we use 1% bovine serum albumin with 10% normal donkey serum). Non-fat dry milk is another option. Non-specific binding of secondary antibody. Use an antibody that has cross-reactive IgG species removed (absorbed against sample species). Tissue dried out. Avoid letting the tissue dry during the staining procedure. Reagents sticking to old or poorly prepared slides. Start over with freshly prepared or purchased slides. Background due to ionic interactions. Increase the ionic strength of the diluent buffer. Problem: Cell/Tissue Morphology is Destroyed
Possible Source Test or Action Antigen retrieval methods are too harsh. Empirically determine the conditions that preserve tissue morphology while restoring the immunoreactivity of the antigen. Tissue sections falling off slide. (more common with frozen sections) Increase fixation time. Empirically determine an additional or alternative fixative.
Use freshly prepared, adequately charged slides.Tissue section appears torn or folded. Air bubbles under section. Re-cut sections using a sharp blade, or ignore damaged areas when analyzing the results. Poor resolution of tissue morphology Cut thinner tissue sections. Ice crystals may have destroyed morphology of frozen sections. Underfixation has physically damaged the tissue or cells during the staining process Increase fixation time and/or add a post-fixation step. Increase the fixative/tissue ratio.
Cut smaller pieces of tissue for more thorough immersion fixation.Autolysis of tissue leading to staining of necrotic debris. Increase the fixation time, ratio. Consider using cross-linking fixative. Problem: Staining is Inappropriate
Possible Source Test or Action Fixation method is inappropriate for the antigen. Try a different fixative or increase the fixation time. Antigen retrieval may be inappropriate for this antigen or tissue. Try different antigen retrieval conditions. Electrostatic charge of the antigen has been altered. Try adjusting the pH or cation concentration of the antibody diluent. Delay in fixation caused diffusion of the antigen. Fix tissue promptly. Try a cross-linking fixative rather than organic (alcohol) fixative.
Our MagCellect Cell Isolation kits are designed to rapidly isolate specific cell populations. We have attempted to make the kits complete and easy to use. To ensure that all users get optimal performance with these products we have summarized some common problems below. We attempt to identify the likely sources of these problems and possible courses of action to be taken to avoid them. We urge users to review these tips and if additional advice is required to contact our technical service department.
Possible Source Test or Action Excessive or nonspecific binding of antibodies and/or ferrofluids to the desired cells Follow the incubation times indicated and/or carry out reactions at the temperature indicated in the product insert Non optimal number of cells used Use the recommended ratio of cells and antibody mixture
Possible Source Test or Action Insufficient antibody used Use the recommended ratio of cells and antibody mixture Non-optimal amounts of ferrofluid used Use the recommended ratio of cells and ferrofluids Carryover of magnetically tagged cells in a negative selection procedure Harvest un-tagged cells carefully without touching the tube wall where magnetically tagged cells are located Magnetic cell separation reaction had not gone to completion Perform the magnetic separation with the recommended magnet and for the recommended time
MycoProbe® Mycoplasma Detection Kit
For further assistance, please contact our technical service department.
Observation: High background level
Problem Corrective Action Insufficient washing Wash per protocol being sure to remove all Wash Buffer from wells before addition of next component Contamination with alkaline phosphatase Keep work area clean and free of alkaline phosphatase Observation: Poor precision
Problem Corrective Action Plate not washed before use Wash per protocol RNase contamination Use RNase-free technique Pipetting error Use a new pipet tip for each pipetting step and use proper technique Observation: No signal for positive control
Problem Corrective Action Component or step omitted Read protocol thoroughly before repeating the assay RNase contamination Use RNase-free technique
Proteome Profiler Array Kits
For further assistance, please contact our technical service department.
Observation Problem Corrective Action No signals on positive control spots Concentration of detection antibody and/or SA-HRP too low Use concentration/dilution specified Chemiluminescent reagents failed Repeat with fresh chemiluminescent reagents according to manufacturers protocol Inadequate exposure time to film Increase exposure time No or low signals on target spots Sample dilution too high Use more sample Analyte abundance in sample is low Use more sample Use different cell line Verify that conditions used to stimulate cells were optimal Sample deteriorated during preparation Supplement buffers with protease and phosphatase inhibitors where indicated Sample deteriorated during storage All samples should be stored at ≤ 70 °C. Avoid freeze-thaw cycles Inadequate exposure time to film Increase exposure time Signals on negative control spots Concentration of detection antibody and/or SA-HRP too high Use concentration/dilution specified Sample concentration too high Use less sample Uneven or high background on blank areas of array Insufficient washing Perform number of washes with volume indicated as specified in the product insert protocol Perform washes in large container and not the 4-Well Multi-dish Concentration of detection antibody and/or SA-HRP too high Use concentration/dilution specified Array was allowed to dry out partially during the procedure Always keep arrays submerged. Minimize time the array is exposed to air. Creased or scratched arrays Use flat-tipped forceps to handle arrays on array number. Too many spots have signals Insufficient washing Perform washes as stated in product insert. Perform washes in a large container, not in the 4-Well Multi-dish. Concentration of detection antibody and/or SA-HRP too high Use concentration/dilution specified Sample concentration too high Use less sample
Proteome Purify™ Immunodepletion Kits
For further assistance, please contact our technical service department.
Problem: Incomplete or no flow through spin filter
Cause Solution Cap on too tight Loosen or remove spin filter cap. Air lock Tap side of spin filter to dislodge air bubble Centrifuge parameters need to be adjusted 2 minutes at 1000-2000 x g usually sufficient.
Western Blot
For further assistance, please contact our technical service department.
- Observation: No Bands Observed
- Observation: Faint Bands (Weak Signal)
- Observation: Extra Bands
- Observation: High Background
- Observation: Diffuse Bands
- Observation: White Bands (ECL method)
- Observation: Patchy uneven spots all over the blot
Observation: No Bands Observed
Possible Source Suggestion Insufficient antibody Antibody may have low affinity to protein of interest. Increase antibody concentration (2-4 fold higher than recommended starting concentration). Antibody may have lost activity. Perform a Dot Blot. Insufficient protein Increase the amount of total protein loaded on gel. Confirm the presence of protein by another method. Use a positive control (recombinant protein, cell line or treat cells to express analyte of interest). Perform a Dot Blot. Poor transfer Wet PVDF/Immobilon-P membrane in methanol or nitrocellulose membrane in transfer buffer. Ensure that there is good contact between PVDF membrane and gel. Incomplete transfer Optimize transfer time. High MW protein may require more time for transfer. To ensure transfer is complete, stain the membrane with Ponceau S, Amido Black or India Ink. Use prestained MW marker (R&D Systems Catalog # MW002 or equivalent). Over transfer Reduce voltage or time of transfer for low molecular weight proteins (< 10 kDa). Isoelectric point is >9 Use alternative buffer system with higher pH such as CAPS (pH 10.5). Incorrect secondary antibody used Confirm host species and Ig type of primary antibody. Old antibody If antibody is expired or past manufacturer warranty, purchase fresh antibody. Incorrect storage of antibodies Follow manufacturer's recommended storage and avoid freeze/thaw cycles. Sodium Azide contamination Make sure buffers do not contain Sodium Azide as this can quench HRP signal. Insufficient incubation time with primary antibody Extend incubation time to overnight at 4°C. Observation: Faint Bands (Weak Signal)
Possible Source Suggestion Low protein-antibody binding Reduce the number of washes to minimum. Reduce NaCl concentration in Blotting Buffer used for wash steps (recommended range 0.15M - 0.5M). Reduce NaCl concentration in Antibody Solution (recommended range 0.15M - 0.5M). Insufficient antibody Antibody may have low affinity to protein of interest. Increase antibody concentration (2-4 fold higher than recommended starting concentration). Insufficient protein Increase the amount of total protein loaded on gel. Inactive conjugate Mix enzyme and substrate in a tube. If color does not develop or, it is weak. Make fresh or purchase new reagents. Switch to ECL. Weak/Old ECL Purchase new ECL reagents. Non-fat dry milk may mask some antigen Decrease milk percentage in Block and Antibody Solutions or substitute with 3% BSA. Observation: Extra Bands
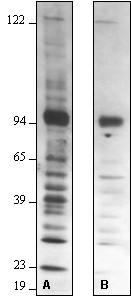
Possible Source Suggestion Non-specific binding of primary antibody Reduce primary antibody concentration. (SeeFigure 1) Reduce the amount of total protein loaded on gel. Use monospecific or antigen affinity purified antibodies (such as R&D Systems "MAB" or "AF" designated antibodies). Non-specific binding of secondary antibody Run a control with the secondary antibody alone (omit primary antibody). If bands develop choose an alternative Secondary Antibody. Use monospecific or antigen affinity purified antibodies (such as R&D Systems "BAF" or "HAF" designated secondary antibodies). Non-specific binding of primary or secondary antibodies Add 0.1 - 0.5% Tween® 20 to primary or secondary Antibody Solution. Use 2% non-fat dry milk in Blotting Buffer as a starting point to dilute primary and secondary antibodies. Adjust antibody concentration up or down as needed. (See Figure 2a & 2b) Increase number of washes. Increase NaCl concentration in Blotting Buffer used for antibody dilution and wash steps (recommended range 0.15M - 0.5M). (SeeFigure 2c) Increase Tween® 20 concentration in Blotting Buffer used for wash steps (0.1%-0.5%). Aggregation of analyte Increase amount of DTT to ensure complete reducing of disulfide bonds (20 -100mM DTT). Heat in boiling water bath 5-10 minutes before loading onto gel. Perform a brief centrifugation. Degradation of Analyte Minimize freeze/thaw cycles of sample. Add protease inhibitors to sample before storage. Make fresh samples. Contamination of reagents Check buffers for particulate or bacterial contamination. Make fresh reagents. Observation: High Background
Possible Source Suggestion Non-specific binding of primary antibody Use monospecific or antigen affinity-purified antibodies (such as R&D Systems "MAB" or "AF" designated antibodies). Block in 5% milk. Adjust the milk (2-5%) or NaCl (0.15-0.5M) concentrations of primary Antibody Solution. (See Figure 3). Decrease antibody concentration. Non-specific binding of secondary antibody Run a control with the secondary antibody alone (omit primary antibody). If bands develop choose an alternative Secondary Antibody. Insufficient blocking Start with 5% dry milk with 0.1%- 0.5% Tween 20, 0.15 -0.5M NaCl in 25mM Tris (pH 7.4). Incubation time may be extended. Adjust milk concentration up or down as needed. Overnight blocking at 4°C may decrease blocking efficiency since detergents might not be effective at lower temperatures. Non-fat dry milk may contain target antigen Substitute with 3% BSA. Non-fat dry milk contains endogenous biotin and is incompatible with avidin/streptavidin Substitute with 3% BSA. Some IgY antibodies may recognize milk protein Substitute with 3% BSA. Insufficient wash Increase number of washes. Increase Tween 20 concentration in Wash Buffer (0.1%-0.5%). Non-Specific Binding of Primary Antibody Increase NaCl concentration in primary Antibody Solution and Blotting Buffer used for dilution of primary antibody and wash steps (recommended range 0.15M - 0.5M). (SeeFigure 4). Film overexposed Reduce exposure time. If target signal is too strong wait 5-10 minutes and re-expose to film. Observation: Diffuse Bands
Possible Source Suggestion Excessive protein on gel Reduce amount of protein loaded Observation: White Bands (ECL method)
Possible Source Suggestion Excessive signal generated Reduce antibody or protein concentration. Excessive antibody or protein can cause extremely high levels of localized signal (usually at a single band). This results in rapid, complete consumption of substrate at this point. Since there is no light production after the completion of this reaction, white bands are the result when exposed to film. Observation: Patchy uneven spots all over the blot
Possible Source Suggestion Contamination of reagents Check buffers for particulate or bacterial contaminate. Make fresh reagents. Not enough solution during incubation or washing Make sure membrane is fully immersed during washes and antibody incubations. Air bubble trapped in membrane Gently remove any air bubbles. Especially during transfer. Uneven agitation during incubations Ensure uniform agitation by placing on a rocker/shaker. Contaminated equipment Make sure that the electrophoresis unit is properly washed. Protein or pieces of gel remaining on the unit may stick to the membrane. Wash membrane thoroughly. HRP aggregation Filter conjugate to remove HRP aggregates. Long exposure Reduce exposure time.
Western Blot Figures
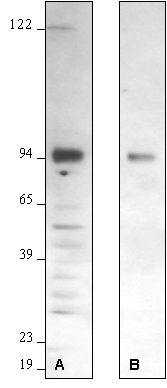
Figure 1 – Effect of Varying Primary Antibody Concentration:
Western blot optimization using R&D Systems rabbit anti-human/mouse/rat RSK1 affinity purified polyclonal antibody (Catalog # AF992). Human HeLa cells were solubilized in SDS gel buffer, and lysate from 1 x 105 cells per lane was resolved by SDS-PAGE. Following electrophoresis, proteins were transferred to an Immobilon-P membrane (Millipore) and immunoblotted under the following conditions. A 30-second exposure to film is shown.
Bolded concentrations indicate the reduction of the primary antibody concentration.
- Blocked in 5% milk, 1.0 microgram/mL primary antibody in 5% milk and 0.15 M NaCl.
- Blocked in 5% milk, 0.2 microgram/mL primary antibody in 5% milk and 0.15 M NaCl.
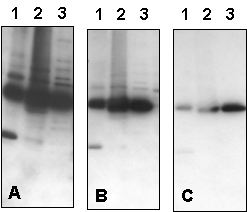
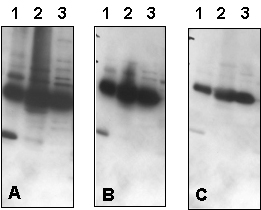
Figure 2 – Effect of Varying Primary Antibody Concentration, % Milk in Antibody Solution, and NaCl Concentration in Blotting Buffer:
Western blot optimization using R&D Systems rabbit anti-human PTP1B affinity-purified polyclonal antibody (Catalog # AF1366). Human HeLa (Lane 1), MCF-7 (Lane 2), and TF-1 (Lane 3) cells were solubilized in SDS gel sample buffer, and lysates from 1 x 105 cells per lane were resolved by SDS-PAGE. Following electrophoresis, proteins were transferred to an Immobilon-P membrane (Millipore) and immunoblotted under the following conditions. A 30-second exposure to film is shown.
Figure 2a – Effect of Varying Primary Antibody Concentration in Antibody Solution with 2% Milk:
Bolded concentrations indicate the dilution of the primary antibody concentration.
- Blocked in 5% milk,1.0 microgram/mLprimary antibody in 2% milk and 0.15 M NaCl.
- Blocked in 5% milk,0.3 microgram/mLprimary antibody in 2% milk and 0.15 M NaCl.
- Blocked in 5% milk,0.1 microgram/mLprimary antibody in 2% milk and 0.15 M NaCl.
Figure 2b – Effect of Varying Primary Antibody Concentration in Antibody Solution with 5% Milk:
In addition to the dilution of the primary antibody concentration (bolded), the milk concentration in the Antibody Solution was also increased.
- Blocked in 5% milk,1.0 microgram/mLprimary antibody in 5% milk and 0.15 M NaCl.
- Blocked in 5% milk,0.3 microgram/mLprimary antibody in 5% milk and 0.15 M NaCl.
- Blocked in 5% milk,0.1 microgram/mLprimary antibody in 5% milk and 0.15 M NaCl.
Figure 2c – Effect of Varying NaCl Concentration in Blotting Buffer:
Bolded concentrations indicate the increased NaCl concentrations in the Blotting Buffer.
- Blocked in 5% milk, 1.0 microgram/mL primary antibody in 5% milk and 0.15 MNaCl.
- Blocked in 5% milk, 1.0 microgram/mL primary antibody in 5% milk and 0.5 MNaCl.
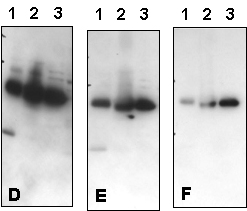
Figure 3 – Effect of Varying Milk and NaCl Concentrations:
Western blot optimization using R&D Systems rabbit anti-human PTP1B affinity-purified polyclonal antibody (Catalog # AF1366). Human HeLa (Lane 1), MCF-7 (Lane 2), and TF-1 (Lane 3) cells were solubilized in SDS gel sample buffer, and lysates from 1 x 105 cells per lane were resolved by SDS-PAGE. Following electrophoresis, proteins were transferred to an Immobilon-P membrane (Millipore) and immunoblotted under the following conditions. A 30-second exposure to film is shown.
Bolded concentrations indicate adjustments in the milk and NaCl concentrations.
- Blocked in 5% milk, 1.0 microgram/mL primary antibody in 2% milk and 0.15 M NaCl.
- Blocked in 5% milk, 1.0 microgram/mL primary antibody in 5% milk and 0.15 M NaCl.
- Blocked in 5% milk, 1.0 microgram/mL primary antibody in 5% milk and 0.5 M NaCl.
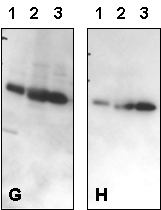
Figure 4 – Effect of Varying NaCl Concentration in Blotting Buffer:
Western blot optimization using R&D Systems rabbit anti-human/mouse/rat RSK1 affinity purified polyclonal antibody (Catalog # AF992). Human HeLa cells were solubilized in SDS gel buffer, and lysate from 1 x 105 cells per lane was resolved by SDS-PAGE. Following electrophoresis, proteins were transferred to an Immobilon-P membrane (Millipore) and immunoblotted under the following conditions. A 30-second exposure to film is shown.
Bolded concentrations indicate an increase in the NaCl concentration in the primary Antibody Solution.
- Blocked in 5% milk, 1.0 microgram/mL primary antibody in 5% milk and 0.15 M NaCl.
- Blocked in 5% milk, 1.0 microgram/mL primary antibody in 5% milk and 0.5 M NaCl.